
Viral Host Predictor
Viral Host Predictor provides a fast and simple way to predict the animal origins of RNA viruses.
Intro
Viral Host Predictor uses the gradient boosting machines algorithms developed by Babayan et al. (2018) to predict the reservoir hosts, arthropod vectors and arthropod-borne transmission status of RNA viruses. Algorithm predictions are based on host-associated biases in viral genomes (‘genomic biases’) and phylogenetic relatedness to viruses with known transmission ecology (“phylogenetic neighbourhood"). Pre-trained machine learning models have been generated using coding and genomic sequences from more than 500 single-stranded RNA viruses in 12 taxonomic groups. Models indicate long-term host associations and do not identify intermediate hosts. Reservoir or arthropod vector groups outside of those shown cannot currently be predicted.
Results are generated by averaging (bagging) predictions across multiple models, each trained and optimised on a different subset of viruses with known host-associations. Across a hold out-dataset these models correctly classified the reservoir hosts of 71.9% of virus species. Accuracy for classifying arthropod vectors was 90.8% and accuracy for classifying viruses that transmit via arthropods from those that do not was 97.9%.
Get Started
Download Example Datasets
These files provide examples of the required input formats for coding and genomic sequences. Each dataset is a zip file containing a pair of coding and genomic sequence FASTA files. Please unzip and then upload the coding file into the coding sequences file input. Optionally, also upload the genome file into the genome sequences file input to run a phylogenetic neighbournood prediction.
Virus Classification
Viral Host Predictor is only intended to be used on sequences of RNA viruses within the following 12 taxonomic groups:
- Arenavirus (family Arenaviridae)
- Astrovirus (family Astroviridae)
- Bunyavirus (order Bunyavirales: families Feraviridae, Hantaviridae, Jonviridae, Nairoviridae, Peribunyaviridae, Phenuiviridae and Tospoviridae)
- Calicivirus (family Caliciviridae)
- Coronavirus (family Coronaviridae)
- Filovirus (family Filoviridae)
- Flavivirus (family Flaviviridae)
- Hepevirus (family Hepeviridae)
- Paramyxovirus (family Paramyxoviridae)
- Picornavirus (family Picornaviridae)
- Rhabdovirus (family Rhabdoviridae)
- Togavirus (family Togaviridae)
Predictions
You can obtain 3 types of prediction from viral sequences:
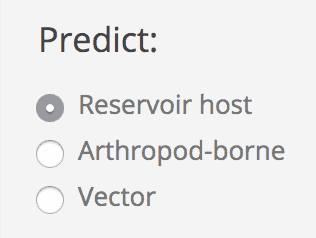
- Reservoir host – the likelihood that the primary host of the virus is a member of any of 12 taxonomic groups
- Arthropod-borne – the likelihood that the virus is either arthropod-borne or has no arthropod vector
- Vector – the likelihood that, if the virus does have an arthropod vector, then the vector is midge, sandfly, mosquito, or tick
You can also select if you want to predict on the basis of genomic biases calculated from coding sequences only or if you want to incorporate the phylogenetic neighbourhood into the prediction, which has been shown to improve the accuracy of results. Both sets of models require a FASTA file containing in-frame coding sequences to be uploaded. Sequences for multiple viruses may be included. Optionally, users can upload an additional FASTA file containing the genome sequences, which will be used for models incorporating the phylogenetic neighbourhood as a predictor.
Options
Predict
Choose which prediction you want to run:
Model
Choose which set of models you want to be used for the prediction:

- Genomic biases + phylogenetic neighbourhood: This option requires for both a coding sequence FASTA file and a genome FASTA file to be uploaded before the Run Predictor button may be clicked. This option predicts with greater accuracy but takes longer as BLAST searches are performed for the genome sequences against multiple BLAST databases.
- Genomic biases: This option requires for a coding sequence FASTA file only to be uploaded before the Run Predictor button may be clicked. Genome sequences are not used and there is no BLAST step. This option produces very fast predictions but predicts with lower accuracy.
Sequence File Formats and Upload
Coding Sequences FASTA File
The virus name is taken from the text on each identifier line, after the > character and up to either the first whitespace, underscore or the end of the line. For example, one virus with 3 coding sequences, and a second virus with a single coding sequence could be entered as follows:
>Virus1 CodingSequence1 ATGGATCCCGGGTAG >Virus1 CodingSequence2 ATGGGGTTTAAATAG >Virus1 CodingSequence3 ATGGATCCGCCGTAG >Virus2 CodingSequence1 ATGCCCGGGGATTAG
Genbank-format identifiers for coding sequences may also be used. In this case, the virus name is the text after >lcl| and before _cds_ or _gene_ in each identifier line.
Viral Host Predictor contains a database of around 3000 Genbank accession numbers along with their associated virus names. If an accession number is supplied instead of the virus name in the identifier line, the accession number is looked up and, if it is found to match an accession number in the database, then the accession number is replaced with the associated virus name.
To summarise, the permitted identifier line formats are as follows:
-
lcl|GenomeAccesion_cds_coding sequence details
-
>GenomeName CodingName
-
>GenomeName_CodingName
- If none of the above is detected, the whole header is treated as a single viral sequence
Multiple coding sequences may be included for each virus. Coding sequences from the same virus must be grouped one after the other in the file. Each sequence must comprise in-frame coding sequence with bases in multiples of 3. N is permitted for an unknown base. U (uracil) is permitted and will be replaced with T (thymine). Any other non-ACGTN letters in sequences will be replaced with N.
Please note that the number of viruses permitted in the coding sequences FASTA file is currently limited to 40 to improve performance.
Genome FASTA File
If a phylogenetic neighbourhood prediction is to be run, a genome FASTA file must be uploaded, with a single genomic sequence corresponding to each virus name in the coding sequences file. For segmented viruses, only the S segment of the genome should be submitted.
The text in each identifier line, after the > character and up to the first whitespace or the end of the line is used as the virus name; each virus name in the coding sequences file must have an exact match in the genome file. Ordering is not important. Additional genome sequences that do not correspond to any coding sequences are permitted but will be excluded from the analysis.
File Upload
The file inputs can accept FASTA files with the following extensions:
- .fasta
- .fna
- .ffn
- .faa
- .fa
- .fst
- .frn
To run a prediction on both genomic biases and phylogenetic neighbourhood (default Model option), you must upload both a coding sequences FASTA file (for calculation of genomic biases) and a genome FASTA (for BLAST searches). The Run Predictor button is activated once both file inputs have been populated.
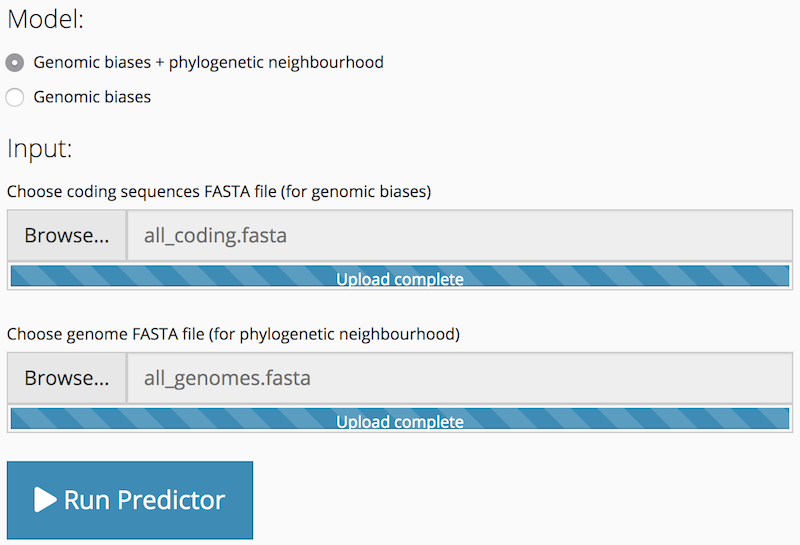
To run a genomic biases only prediction, after selecting the Model: Genomic biases option, you only need to upload a coding sequences FASTA file. The genome FASTA file input is hidden. The Run Predictor button is activated after the coding sequences FASTA file input has been populated.
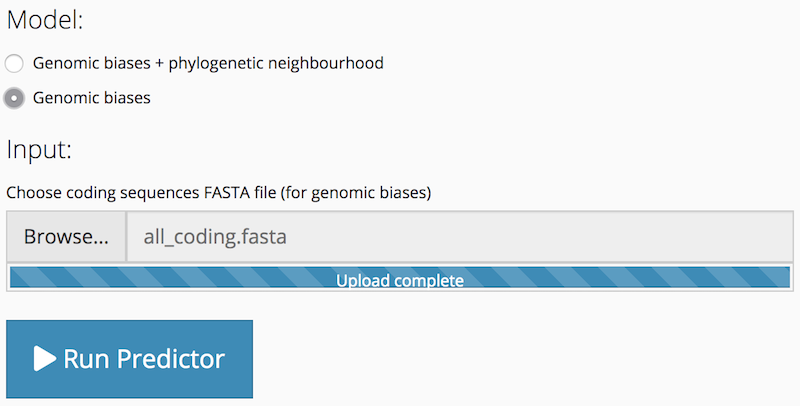
Run Predictor
After you have chosen options and uploaded sequence data, click the Run Predictor button to start the prediction process. The overall length of time the prediction takes varies according to the amount of sequence data; in particular, a lot of genomic data will increase the time taken for BLAST searches. The application provides you with continuous feedback about the progress of the prediction process via the progress indicator that is displayed in the bottom right-hand corner of the window.
The prediction process has 2 or 3 main steps:
- Calculation of genomic biases
- BLAST searches (for phylogenetic neighbourhood predictions)
- Machine learning predictions
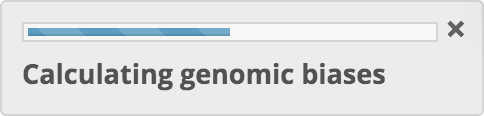
Calculation of genomic biases
Sequence features, ranging from nucleotide and amino acid frequencies, to dinucleotide, codon and codon pair biases, are calculated from the coding sequences FASTA file. The features that have been determined to be most predictive for the type of prediction being run are selected.
A progress indicator is displayed. The coding sequences FASTA file is then pre-processed and validated. For details of pre-processing, see the section Coding Sequences FASTA File above. If any > characters are found within sequences, or if any sequences have fewer than 3 characters, processing stops and an error message is displayed.
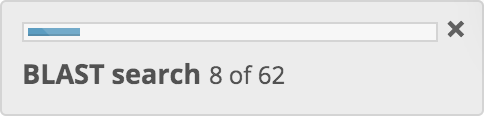
BLAST searches (optional)
If a phylogenetic neighbourhood prediction has been chosen, BLAST searches are then performed for sequences in the genome FASTA file against multiple BLAST databases. These BLAST databases have been generated from genomic sequences in the training data sets that have been shown to generate most accurately predictive machine learning models. The BLAST searches may take a few minutes to complete.
The genome FASTA file is validated before proceeding with BLAST searches. The sequence names in the genome FASTA file are cross-referenced against the names in the coding sequences FASTA file. If any sequence is found in the coding sequences FASTA file that does not correspond to a sequence in the genome FASTA, or if any duplicate sequence names are found in the genome FASTA, processing stops and an error message is displayed.
If any sequences are found in the genome FASTA file without any corresponding sequences in the coding FASTA file, these genomic sequences are removed before proceeding with BLAST searches. These redundant sequence names are displayed in a warning notification.
A progress indicator is displayed and is continuously updated as the BLAST searches progress.
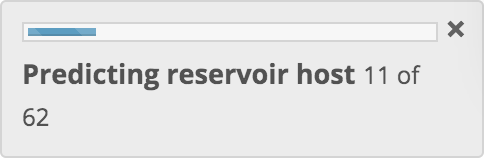
Machine learning predictions
Following data preparation, genomic features (and optionally BLAST hits) are used to generate predictions of reservoir host, arthropod-borne vector status or vector. The most predictive models are selected for the chosen prediction and a prediction is run against each of those models in turn. A progress indicator is displayed and is updated following each prediction. Results from these multiple predictions are then combined.
Results
Multiple View
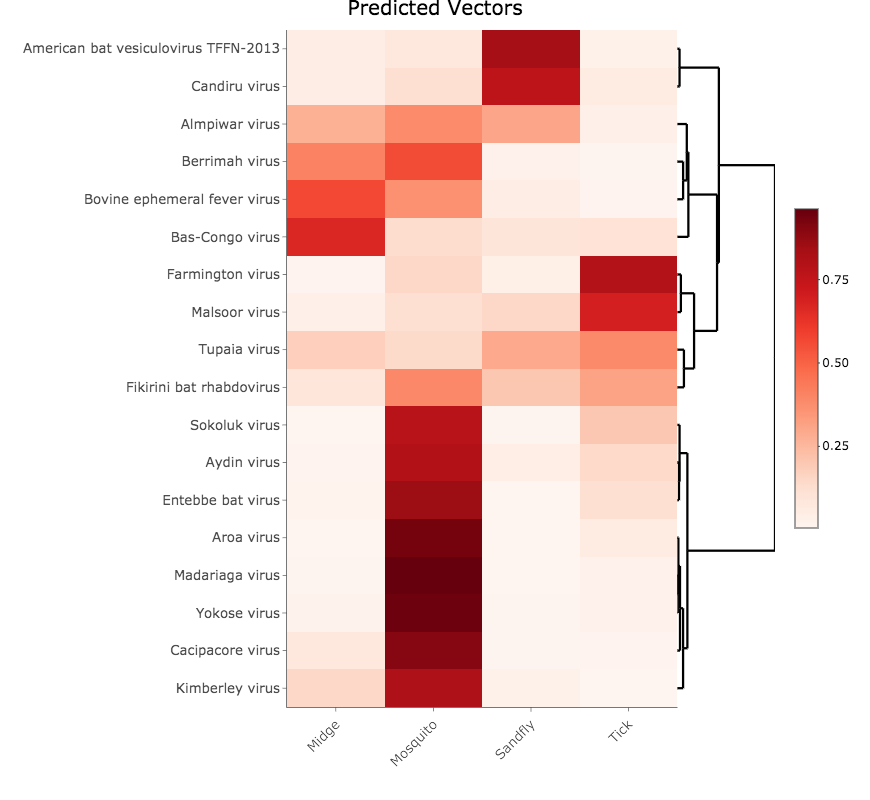
If your coding sequences contain multiple sample names, you may choose to visualise mean predicted probabilities for all samples together in a heatmap.
An interactive heatmap is displayed in the main panel of the application. You can hover the mouse over an individual cell to view the mean probability value for that sample across all predictions for each classification. To download a screenshot as a PNG file, click the camera icon that appears in the top right corner when you hover the mouse over the heatmap. To download a table of all averaged probability values, click the Download Results button.
Single View
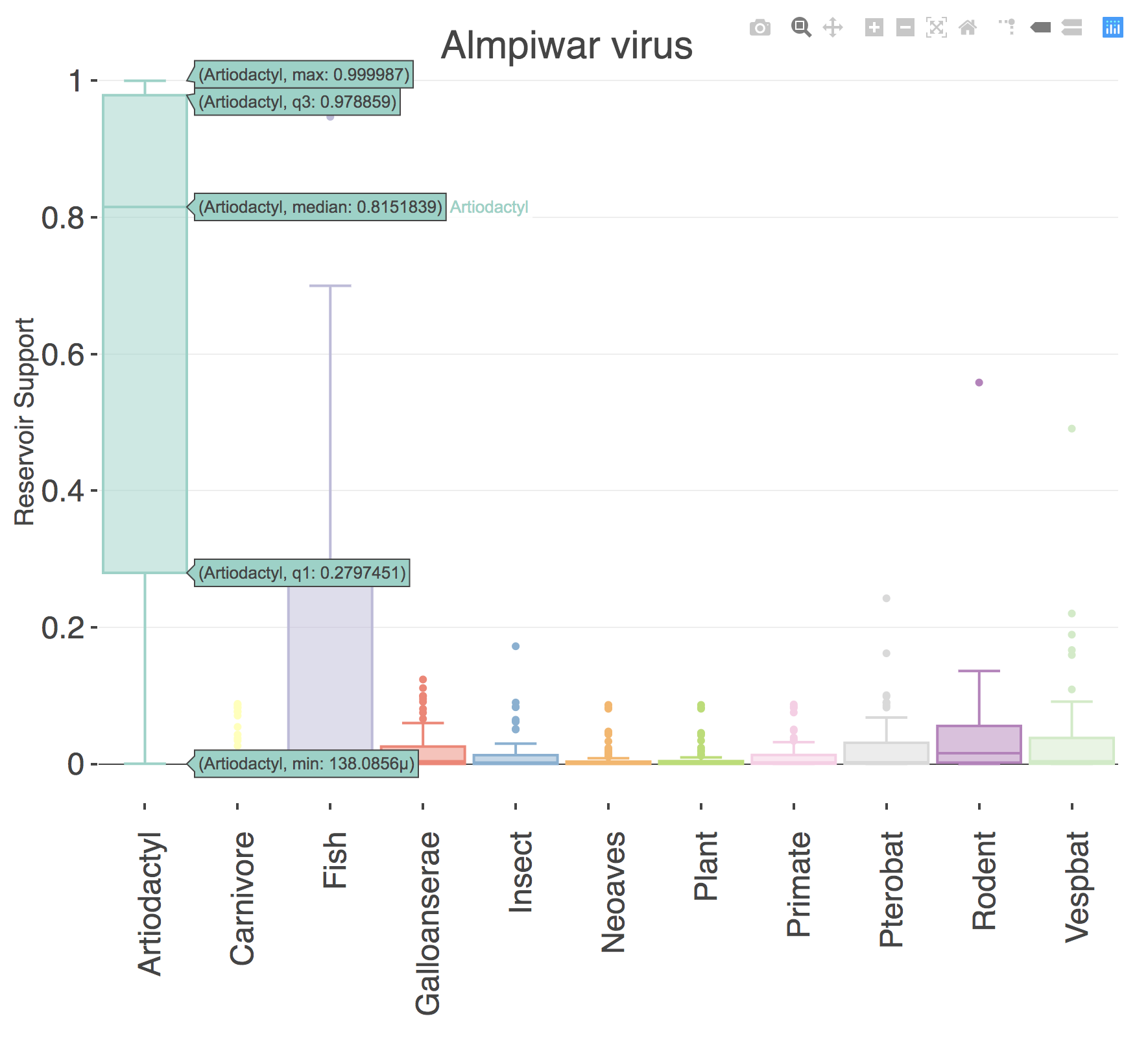
You can also select a single sample to visualise individually as a boxplot, showing the range of predicted probability values across all machine learning models for each classification. N.B. If your coding sequences contain only a single sample name, plot options are not displayed and a boxplot is displayed for the individual sample.
An interactive boxplot is displayed in the main panel of the application. You can hover the mouse over an individual box to view the statistical values in the distribution of probabiilties across all machine learning models for that classification. To download a screenshot as a PNG file, click the camera icon that appears in the top right corner when you hover the mouse over the boxplot. To download a table of all prediction values in the boxplot, click the Download Results button.
References
Citation
Please cite:
Machine Learning Models
The source code used to train the machine learning models in Viral Host Predictor may be downloaded from GitHub.
Images
- SARS-CoV-2 cutaway pleomorphic by Nick Woolridge / Biomedical Communications. Creative Commons Attribution-NoDerivatives 4.0 International License.
- An anopheles mosquito (Aedes aegypti). Coloured drawing by A.J.E. Terzi. Wellcome Collection. Attribution 4.0 International (CC BY 4.0).
Contact
Please send your comments, suggestions or bug reports to Derek Wright.